ATUALIZAÇÃO NO CHOQUE VASOPLÉGICO.
PARTE I. BASES FISIOPATOLÓGICAS

I.
INTRODUÇÃO
A
edição de março do Critical Care nos trouxe uma interessante revisão de Wieruszewski,
PM e Khanna, AK, sobre o uso de vasopressores no choque circulatório de
tipo vasoplégico. Trata-se de uma das dez revisões selecionadas do Annual
Update in Intensive Care and Emergency Medicine 2022, da série BioMedical
Center (BMC) e publicada na revista Critical Care em 22 de março de
2022[1]. O
enfoque dessa revisão é discutir o uso da terapia vasopressora no choque
vasoplégico, com ênfase no que a literatura em vigor versa a respeito do
momento do início (precoce versus tardio) da terapia vasopressora usando a norepinefrina
como droga de escolha inicial; o momento em que se deveria associar uma segunda
droga vasopressora adjuvante e qual seria ela. Ainda, os autores defendem que,
considerando os mecanismos fisiopatológicos do choque vasoplégico, suas repercussões
hemodinâmicas (progressão para choque refratário, irreversível e finalmente
óbito) e a necessidade de usar doses elevadas de norepinefrina, muitas vezes
gerando reações adversas graves, se faz necessário pensar numa TERAPIA VASOPRESSORA
MULTIMODAL PRECOCE E DE AMPLO ESPECTRO (a semelhança da antibioticoterapia
empírica inicial na sepse e choque séptico) visando atuar simultaneamente nos diferentes
mecanismos fisiopatológicos do choque vasoplégico, que permitam uma reversão
mais rápida do choque evitando-se os possíveis efeitos adversos de uma
monoterapia com norepinefrina em doses elevadas.
É
com base nessa revisão que agora pretendemos ampliar o debate a respeito dessa
estratégia que parece realmente plausível, apesar de que ainda faltam estudos
que, do ponto de vista da medicina baseada em evidências, garantam realmente
benefícios e ausência de malefícios ao paciente. Para tanto, teceremos algumas
considerações a respeito dos mecanismos fisiopatológicos envolvidos no choque
vasoplégico e sua progressão e finalmente abordaremos as questões pertinentes à
estratégia de terapia vasopressora multimodal precoce e de amplo espectro.
Tomando
como referência uma revisão no UpToDate[2]
encontramos que o choque circulatório (ou simplesmente choque) é
definido como um estado de insuficiência circulatória que leva a hipóxia
celular e tecidual, seja por queda na oferta de oxigênio (DO2), aumento do
consumo de oxigênio (VO2), utilização inadequada de oxigênio ou uma combinação
desses processos. Ocorre mais comumente quando há insuficiência circulatória
manifestada como hipotensão (isto é, perfusão tecidual reduzida). Entretanto,
um paciente em choque pode apresentar-se hipertenso, normotenso ou hipotenso. O
choque é inicialmente reversível, mas deve ser reconhecido e tratado
imediatamente para evitar a progressão para o estado de irreversível. Usualmente
o choque circulatório é classificado em 4 grandes grupos: distributivo,
cardiogênico, hipovolêmico e obstrutivo, podendo cada um deles se subdividir em
subgrupos de acordo com a sua etiologia. Muitos pacientes com insuficiência
circulatória apresentam uma combinação de mais de uma forma de choque (choque
multifatorial ou misto). Choque indiferenciado, refere-se à situação em
que o choque é reconhecido, mas a causa não é clara ou é desconhecida.
Assim,
tomando como referência o UpToDate podemos classificar o choque como:
1.
DISTRIBUTIVO (VASOPLÉGICO).
1.1
SEPTICO: De acordo com o agente etiológico:
1.1.1
Por Gram positivos: pneumococcus, staphylococcus, streptococcus, enterococcus, listeria)
1.1.2.
Por Gram negativos: klebsiella, pseudomonas, escherichia, haemophilus, legionella, neisseria,
moraxella, rickettsia, francisella (tularemia)
1.1.3
Por Fungos: candida, aspergillus.
1.1.4.
Por Virus: influenza, citomegalovírus, ebola, varicela.
1.1.5.
Por parasitas: plasmodium, ascaris, babesia.
1.1.6.
Por Micobacterias: mycobacterium tuberculosis, mycobacterium abscessos.
1.2
NÃO SÉPTICO:
1.2.1.
Choque inflamatório ou sirético (decorrente de síndrome da resposta
inflamatória sistêmica - SIRS): queimaduras, trauma, pancreatite,
pós-infarto do miocárdio, pós by-pass coronário, pós parada cardíaca,
perfuração de vísceras, embolia de líquido amniótico, embolia gordurosa,
síndrome de vazamento capilar sistêmico idiopático.
1.2.2.
Choque neurogênico: traumatismo cranioencefálico, lesão medular (quadriparesia com
bradicardia ou paraplegia com taquicardia), anestesia neuraxial.
1.2.3.
Choque anafilático: mediado por IgE (exemplo, desencadeado por comidas, mordidas de
cobra ou picadas de insetos incluindo envenenamento por escorpião e várias
picadas de aranha), não mediadas por IgE (exemplo, ferro, dextrans), não
imunológico ou imunomediado (exemplo, induzido por exercício ou calor, meios de
contraste, látex etc.), idiopático.
1.2.3.
Outros: pós-operatório (pós circulação extracorpórea - CEC), insuficiência
hepática, reações transfusionais, induzido por drogas (por exemplo, agentes
vasodilatadores, induzido por toxinas, (por exemplo, metais pesados, incluindo
arsênico, ferro e tálio), infecções associadas à síndrome do choque tóxico (por
exemplo, Streptococcus e Escherichia), beribéri.
2.
CARDIOGÊNICO.
2.1
CARDIOMIOPÁTICO: Infarto do miocárdio (envolvendo >40% do ventrículo esquerdo
ou com isquemia extensa), infarto grave do ventrículo direito, exacerbação
aguda de insuficiência cardíaca grave por cardiomiopatia dilatada, miocárdio
atordoado por isquemia prolongada (por exemplo, parada cardíaca, hipotensão,
circulação extracorpórea), choque séptico avançado (miocardiopatia séptica), miocardite,
contusão do miocárdio, induzida por drogas (por exemplo, betabloqueadores)
2.2
ARRTIMOGÊNICO
2.2.1
Taquiarritmias: taquicardias atriais (fibrilação, flutter, taquicardia
reentrante), taquicardia ventricular e fibrilação ventricular.
2.2.2
Bradiarritmias: bloqueio cardíaco completo, bloqueio cardíaco de segundo grau
Mobitz tipo II.
2.3
MECÂNICO
Insuficiência
valvar grave, ruptura valvar aguda (ruptura papilar ou de corda tendinosa ou
cordoalha, abscesso valvar), estenose valvar crítica, defeito da parede do
septo ventricular agudo ou grave, aneurisma da parede ventricular roto, mixoma
atrial.
3.
HIPOVOLÊMICO
3.1
HEMORRÁGICO
Trauma,
hemorragia digestiva (exemplo, sangramento de varizes de esôfago, úlcera
péptica), hemorragia intra ou pós-operatória, hemorragia retroperitonial
(exemplo, rotura de aneurisma de aorta), fístula aorto-entérica, pancreatite
hemorrágica, iatrogênica (exemplo, lesão inadvertida de malformação
arterio-venosa ou de ventrículo esquerdo, erosão de tumor ou abscesso em
grandes vasos, gravidez ectópica rota, hemorragia pós-parto, hemorragia uterina
ou vaginal (exemplo, infecção, tumores, lacerações), hemorragia peritoneal
espontânea por diátese hemorrágica.
3.2
NÃO HEMORRÁGICO
Perdas
gastrointestinais (por exemplo, diarreia, vômito, drenagem externa); perdas por
pele (por exemplo, insolação, queimaduras, condições dermatológicas); perdas
renais (por exemplo, diurese osmótica ou induzida por drogas, nefropatias
perdedoras de sal, hipoaldosteronismo); perdas para o terceiro espaço, para o
espaço extravascular ou cavidades corporais (por exemplo, pós-operatório e
trauma, obstrução intestinal, lesão por esmagamento, pancreatite, cirrose).
4.
OBSTRUTIVO
4.1
VASCULAR PULMONAR: embolia pulmonar com instabilidade hemodinâmica, hipertensão
pulmonar grave, obstrução grave ou aguda da válvula pulmonar ou tricúspide,
embolia gasosa venosa.
4.2
MECÂNICO: pneumotórax hipertensivo ou hemotórax volumoso (p. ex., trauma,
iatrogênico), derrame pericárdico com tamponamento cardíaco, pericardite
constritiva, cardiomiopatia restritiva, hiperinsuflação dinâmica grave (exemplo,
níveis elevados de PEEP intrínseca), obstrução da via de saída do ventrículo
direito ou esquerdo, síndrome compartimental abdominal (SCA), compressão
aorto-cava (exemplo, posicionamento, retração cirúrgica).
5.
MISTO (MULTIFATORIAL)/DE ETIOLOGIAS DIVERSAS.
5.1
Endócrino: por exemplo, insuficiência adrenal, tireotoxicose, coma mixedematoso.
5.2
Metabólico: por exemplo, acidose, hipotermia.
5.3
Outros: politrauma com mais de uma categoria de choque, etiologia de
choque agudo com doença cardíaca pré-existente, choque tardio sub-ressuscitado,
intoxicações diversas. O cianeto e o monóxido de carbono causam choque por
disfunção mitocondrial.
IMPORTANTE:
Os
pacientes geralmente apresentam formas mistas de choque, seja desde sua
admissão numa unidade de emergência ou durante sua evolução. Exemplos:
·
Pacientes com choque séptico ou, sirético por pancreatite,
apresentam choque principalmente distributivo (vasoplégico), levando a um
componente hipovolêmico relativo, devido aos efeitos produzidos pelas citocinas
pró-inflamatórias e anti-inflamatórias na permeabilidade vascular, gerando
vasodilatação e redistribuição da volemia com redução do volume circulante efetivo.
No entanto, eles também costumam ter um componente hipovolêmico real (devido à
diminuição da ingestão oral, perdas insensíveis, vômitos, diarreia) e um
componente cardiogênico (devido à depressão miocárdica relacionada à
inflamação).
·
Pacientes com cardiomiopatia subjacente podem apresentar choque
hipovolêmico (por excessiva diurese forçada por drogas) e choque cardiogênico
(por taquicardias compensatórias que ao encurtar a diástole levam a queda do
volume sistólico).
·
Pacientes com lesão traumática grave podem ter choque hipovolêmico
hemorrágico por perda de sangue, bem como choque distributivo por SIRS ou,
menos comumente, choque obstrutivo por TEP ou embolia gordurosa.
·
Pacientes com trauma na medula espinhal podem apresentar choque
distributivo (vasoplégico) por disfunção autonômica relacionada a inibição
simpática e choque cardiogênico por depressão miocárdica.
·
Pacientes com ruptura de aneurisma de parede livre do ventrículo
esquerdo podem apresentar choque cardiogênico por falha primária da bomba, choque
obstrutivo por tamponamento cardíaco quando a perda de sangue é contida pelo
saco pericárdico e choque hemorrágico quando a perda de sangue não é contida
pelo saco pericárdico.
·
Pacientes com choque séptico podem fazer a transição de um estado
de choque distributivo (baixa resistência vascular sistêmica) para um estado de
choque multifatorial após ressuscitação com volume maciço que resulta em
síndrome compartimental abdominal e/ou insuficiência cardíaca aguda do coração
direito.
Por
outro lado, diante de um estado de choque que se inicia, o desequilíbrio
homeostático se produz por diferentes mecanismos que aparecem simultaneamente
ou de forma sequencial em cada tipo de choque. Assim, a restauração do equilíbrio
homeostático deveria ter como alvos tais mecanismos de forma simultânea ou
sequencial (terapia multimodal do choque). Para tanto, se faz importante
entender como ocorrem tais mecanismos em cada tipo de choque.
O
CHOQUE DISTRIBUTIVO (VASOPLÉGICO), é a forma mais comum de choque
circulatório encontrado em pacientes internados na unidade de terapia intensiva
(UTI), seguido pelo choque cardiogênico[3], e
a sepse sua etiologia predominante (choque vasoplégico de causa infecciosa).
Entretanto, como visto acima, outras causas não associadas à infecção podem
levar a intensa vasoplegia e choque. Mesmo breves períodos de hipotensão
no período intraoperatório podem levar a lesão renal e miocárdica[4].
O
choque vasoplégico é uma emergência médica que requer diagnóstico e tratamento imediatos. Independentemente
da sua etiologia, é caracterizado pela redução da resistência vascular
sistêmica (RVS) que leva a hipotensão arterial, sendo necessário o manejo com
ressuscitação volêmica intravascular e uso de drogas vasopressoras para
restaurar o tônus vascular. Sem tratamento, as pressões de perfusão se
reduzem para níveis severos/críticos, levando à utilização inadequada de
oxigênio celular, desvio para metabolismo anaeróbico (com queda drástica na
produção de ATP como fonte de energia), falência de múltiplos órgãos e morte[5] [6].
III.
TÔNUS VASCULAR E RESISTÊNCIA VASCULAR E PRESSÃO ARTERIAL.
Sabemos
que a pressão arterial (PA) está diretamente relacionada ao débito cardíaco
(DC) e à resistência vascular sistêmica (RVS). Ou seja:
PA = DC X RVS.
A
pressão arterial é regulada a curto prazo por sistemas de barorreceptores,
quimioreceptores e receptores cardiopulmonares (neurônios especializados no
arco aórtico e seios carotídeos), cujas aferências se projetam para o sistema
nervoso central, especificamente ao núcleo do trato solitário (NTS) via nervos
vagos e glossofaríngeo. O processamento dessas informações no sistema nervoso
central produz uma regulação das vias autonômicas eferentes, havendo, assim, o
controle da distribuição do débito cardíaco em diferentes leitos vasculares.
Este evento fornece um feedback negativo em que a pressão arterial elevada
reflexivamente provoca diminuição da pressão. Em contraste, a diminuição da
pressão arterial deprime o barorreflexo, fazendo com que a pressão arterial
aumente. Em níveis normais de pressão arterial, os barorreceptores inibem
tonicamente os efeitos simpáticos sobre os vasos sanguíneos e coração[7]. O
controle da PA a longo prazo é exercido pelo sistema
renina-angiotensina-aldosterona (SRAA) com a regulação do volume dos líquidos
corporais (volemia), regulando o equilíbrio hidroeletrolítico do corpo.
A
resistência vascular sistêmica (RVS) é influenciada em graus variados,
dependendo da região, pela atividade de nervos simpáticos vasomotor, pelo nível
de hormônios vasoativos circulantes e por fatores locais, incluindo fatores
metabólicos produzidos pela camada endotelial[8]. Assim,
a hipotensão arterial decorrente de vasoplegia é consequência da queda da RVS
que em última instância traduz uma perda do “tônus vascular”.
1.
TÔNUS VASCULAR OU VASOMOTOR. O tônus vascular, é um
termo comumente usado para caracterizar o estado contrátil de das fibras
musculares lisas que fazem parte da camada média de um vaso seja arterial ou
venoso, determinado assim o seu diâmetro. Sabe-se que as arteríolas são os
principais vasos que determinam resistência vascular sistêmica (é o vaso que
proporcionalmente tem a maior camada muscular lisa). A musculatura lisa que se
organiza de forma circular em torno desses vasos é a responsável pelo tônus que
regula a luz do sistema arterial. O
tônus basal, refere-se ao estado de constrição parcial mesmo quando
todas as influências externas sobre elas são removidas (às vezes chamado de tônus
intrínseco). A compreensão do mecanismo é incompleta, mas o tônus arteriolar
basal pode ser um reflexo do fato de que as células musculares lisas resistem
inerente e ativamente ao alongamento decorrente de estar continuamente
pressurizadas. Outra hipótese, é que o tônus basal das arteríolas seja
resultado de uma produção tônica de substâncias vasoconstritoras pelas células
endoteliais que revestem sua superfície interna. De qualquer forma, esse tônus
basal estabelece um “estado basal de constrição arteriolar parcial” a partir
do qual as influências externas nas arteríolas exercem seus efeitos dilatadores
ou constritores[9].
2.
REGULAÇÃO DO TÔNUS VASCULAR. A regulação do tônus vascular
se dá de 2 formas: intrínseca e extrínseca.
2.1
REGULAÇÃO INTRÍNSECA, se dá por fatores ou mecanismos do próprio endotélio vascular, em
resposta a estímulos mecânicos (forças de cisalhamento) e farmacológicos. Nesta
forma de regulação, o endotélio se comporta como se fosse um “tecido
endócrino”, já que sintetiza e secreta substâncias que provocam contração
(vasoconstrição) ou relaxamento (vasodilatação) do músculo liso vascular que
rodeia o endotélio, em resposta a forças ou estímulos[10]. Estas
substâncias agem principalmente provocando aumento ou diminuição do cálcio
intracelular (Ca⁺⁺) levando respectivamente a vasoconstrição ou
vasodilatação.
Existem
forças que atuam na parede do vaso sanguíneo e que são criadas pelo fluxo
sanguíneo e a energia criada pela contração ventricular esquerda. Estas forças são
basicamente de dois tipos. Uma é uma força decorrente da pressão (P) do sangue que
atua verticalmente sobre a parede do vaso e reflete os efeitos da pressão
arterial em si. A outra força exercida pelo fluxo sanguíneo atua paralelamente
ou tangencialmente sobre a parede do vaso, sendo denominada tensão de
cisalhamento ou shear stress (W). A Fig.1 mostra o sentido das forças.

Fig. 1 Forças que atuam sobre o
endotélio vascular
Ambas
as forças, principalmente as de cisalhamento atuam diretamente sobre o
endotélio vascular desencadeando uma resposta celular endócrina da célula
endotelial com produção de substâncias que exercem efeitos vasoconstritores e
vasodilatadores nas células muscular lisas que as rodeiam. São forças transmembrana que se
relacionam diretamente com a membrana nuclear, que ao sofrer alterações causam
mudanças na expressão de proteínas, bem como através do citoesqueleto, e
alteram o modo como as células operam em relação a outras células às quais se
opõem[11].
Além
das forças mecânicas acima descritas, o endotélio vascular responde também a estímulos
farmacológicos extrínsecos como: autacóides (bradicinina-BK), hormônios
circulantes (angiotensina II), neurotransmissores (acetilcolina, noradrenalina)
e ainda produtos derivados de plaquetas e coagulação (serotonina, trombina)
sendo responsável pela ativação de diferentes enzimas produzindo diferentes
efeitos vasoativo[12]. Metabólitos
secretados durante a realização de exercício físico como lactato, adenosina e
CO2, também promovem vasodilatação. A histamina secretada pelos mastócitos e
basófilos, promove intensa vasodilatação durante uma reação alérgica. Em
oposição às substâncias vasodilatadoras, temos o tromboxano A2 e a serotonina
no grupo de agentes vasoconstrictores. Nestas situações, o endotélio faz a
regulação intrínseca do tônus vascular. Isto é feito através de um equilíbrio
na secreção de substâncias vasodilatadoras (como o óxido nítrico) e
vasoconstritoras (como a endotelina-1). Além disso, o endotélio vascular possui
uma ampla variedade de receptores para várias substâncias que atuam na
regulação do tônus vascular, dentre elas a insulina, angiotensina II e
prostaglandinas. Como veremos mais adiante, a regulação extrínseca direta
provoca aumento do tônus simpático promovendo maior secreção de catecolaminas
(adrenalina e noradrenalina) da zona medular da glândula adrenal, sendo
predominante a secreção de adrenalina. A adrenalina tem, em doses baixas,
efeito predominante β (β1 inotrópico e β2 vasodilatador) e em doses altas efeito
α (vasoconstritor). Já a noradrenalina, também da adrenal, possui apenas o
efeito vasoconstrictor α. O sistema parassimpático não possui ação
vasodilatadora generalizada, e atua sobre alguns órgãos (exemploj: glândulas
salivares, órgãos genitais, etc.). O hormônio antidiurético ou vasopressina
possui um efeito vasoconstritor sendo secretado em resposta a um aumento da
osmolaridade sanguínea[13].
As
principais substâncias vasoconstritoras endoteliais são:
a) Substâncias derivadas do ácido araquidônico
(AA) sintetizadas pela ação da ciclooxigenase (COX): tromboxanos
(TXA2) e prostaglandinas (PGH2). Induzem a ativação via proteína G-fosfolipase
C (PLC) e aumento do cálcio, levando à contração da célula muscular;
b)
Espécies reativas de oxigênio (ROS). Radicais livres são átomos ou
moléculas que, devido a apresentarem um ou mais elétrons desemparelhados em
suas camadas de valência, apresentam forte tendência a oxidar (ceder os
elétrons desemparelhados) outras moléculas, que ao recebê-los ficam reduzidas. Emparelhamento
ou desemparelhamento de elétrons se refere ao spin dos elétrons nas orbitais
(spin: define o sentido da rotação dos elétrons). Ou seja, em cada orbital
podemos ter no máximo 2 elétrons com spin oposto, um com +1/2 e outro com -1/2
(princípio da exclusão de Pauli). Elétrons emparelhados significa que estão aos
pares nas orbitais com seus spins no mesmo sentido. Elétrons desemparelhados
significa que não têm o seu par na orbital. O oxigênio molecular (O2) é um dos
radicais mais conhecidos do nosso organismo, pois em seu estado estável
apresenta dois elétrons desemparelhados nos orbitais antiligantes, sendo um
potente agente oxidante. ROS são formas reduzidas de oxigênio (receberam mais
elétrons) que são energeticamente mais reativas que o oxigênio molecular, ou
seja, são compostos que tem maior facilidade em reagir com outras substâncias
podendo gerar uma cascata de reações. As formas mais comuns de ROS encontrados
nas células são: radicais superóxido (O2–), hidroxila (OH–), peróxido de
hidrogênio (H2O2) e oxigênio singleto (¹O2). O O2 tem forte tendência de
receber somente um elétron por vez devido à sua configuração eletrônica,
formando o radical superóxido (O2–). Se receber mais um elétron e dois íons hidrogênio,
há formação de peróxido de hidrogênio (H2O2). Por sua vez, se o H2O2 receber
mais um elétron e um íon hidrogênio, forma-se o radical hidroxila (OH–). O
oxigênio singleto (¹O2) apresenta dois elétrons emparelhados que podem estar
num mesmo orbital ou em orbitais diferentes. Os radicais superóxido (O2–), podem
reagir com o oxido nítrico (NO) para formar peróxidos de nitrito (ONOO-),
contribuindo para a diminuição da disponibilidade desse potente vasodilatador
endotelial induzindo assim vasoconstrição. O oxigênio singleto (¹O2) parece também
estar envolvido na formação de uma molécula sinalizadora que regula o tônus
vascular e a pressão sanguínea durante a inflamação sistêmica aguda.
c)
Endotelina-1 (ET-1). Vasoconstrictor
ativador do subtipo de receptor para endotelina A (ETA) ou B (ETB), em células
musculares vasculares. A ET-1 leva à ativação de receptores acoplados a
proteína G, predominantemente, Gq/11 e G12 levando a vias de mobilização do
cálcio intracelular e ativação de vias dependentes da RhoA/ Rho-cinase. No
entanto, a ativação dos receptores ETB presentes na célula endotelial pode
levar à ativação de fatores relaxantes derivados do endotélio.
As
principais substâncias vasodilatadoras endoteliais são:
a)
Sustâncias derivadas do ácido araquidônico (AA) sintetizados pela ação da
ciclooxigenase (COX): prostaglandinas (PGI2); da lipoxigenase: ácido
15-s-hidroxieicosatetraenóico (15-HETE) e do citocromo P450 (CYP450). PGI2 aciona
receptores ligados à proteína Gs, células musculares lisas, e ativam a adenilciclase
resultando no aumento de AMPc que, via PKA, induz o relaxamento muscular liso.
b)
Fator hiperpolarizante derivado do endotélio (EDHF). Sua
formação pode ser dependente da sinalização do cálcio intracelular, induzida
pela ativação de receptores acoplados à proteína G. No entanto, a natureza do
fenômeno EDHF é questionada. Sabe-se apenas que ocorre uma hiperpolarização
dependente da ativação de canais de potássio dependentes do cálcio com média e
pequena condutância localizados nas células endoteliais. A hiperpolarização
endotelial é um pré-requisito para a hiperpolarização e relaxamento de células
musculares lisas vasculares. A transmissão da hiperpolarização entre o
endotélio e músculo liso vascular, produzida por junções tipo gap
mioendoteliais, implica na ativação de outros canais de potássio, como os
canais retificadores de entrada, e ativação da bomba Na+/K+ ATPase, aumentando
o estado hiperpolarizado da célula. Esta hiperpolarização é o principal efeito
que produz o fechamento dos canais de cálcio sensíveis à voltagem (CaV), ocorrendo
diminuição da concentração de cálcio intracelular e relaxamento da musculatura
lisa vascular. Atualmente, várias hipóteses são descritas para determinar a
natureza estrutural e bioquímica do EDHF, entre elas os metabólitos da enzima
CYP450, ácidos epoxieicosatrienóicos (EET), íons potássio (K+), peróxido de
hidrogênio (H2O2) e radicais nitroxil (HNO).
c) Oxido nítrico (NO).
Identificado pela primeira vez como o fator relaxante derivado do endotélio
(EDRF) é talvez o mais importante vasodilatador que desempenha um papel de
extrema relevância não apenas no controle fisiológico do tônus vascular, mas na
fisiopatologia do choque vasoplégico de diferentes etiologias. Trata-se de um radical livre, gasoso,
inorgânico, incolor, com uma meia vida relativamente curta cuja síntese endotelial
se dá pela conversão enzimática, mediada pela oxido nítrico sinetasse (NOS), do
aminoácido L-arginina, em duas etapas. A primeira é uma N-hidroxilação do grupo
guanidina da L-arginina, formando um intermediário N-hidróxi-L-arginina
(L-NOHA) na presença de O2 e cofatores essenciais como NAD(P)H e
(6R-)5,6,7,8-tetrahidro-L-biopterina (BH4). Na segunda etapa, L-NOHA é
metabolizada em L-citrulina e NO (Fig. 2).

Fig.2 Síntese do Oxido Nítrico
(NO)
Existem
3 isoformas de NOS: a óxido nítrico sintetase neuronal (nNOS), a induzível
(iNOS) e a endotelial (eNOS), sendo que nas células do endotélio vascular foram
caracterizadas a eNOS e a iNOS. A isoforma endotelial (eNOS) é um determinante
chave da homeostase vascular e é regulada através de diversos receptores de
superfície celular. Devido à sua natureza lipofílica, o NO produzido difunde-se
através das células da musculatura lisa adjacentes, e pode atuar ligando-se de
forma reversível ao grupo heme da guanidil ciclase (GC) ativando-a, transformando
a guanosina trifosfato (GTP) em guanosina monofosfato cíclico (GMPc), que por
sua vez, ativa a proteína cinase G (PKG) promovendo a recaptação de Ca2+ para
os estoques intracelulares, via bomba de cálcio (SERCA), abertura dos canais de
K+; fechamento dos canais de Ca2+. Todos estes eventos levam à redução da
concentração de cálcio intracelular e relaxamento vascular. No entanto, autores
propuseram que o NO poderiam ativar diretamente canais para K+ do músculo liso
vascular, independente da ativação da GC solúvel, levando a hiperpolarização e
ao relaxamento vascular.
O
aumento do Ca2+ no citoplasma (gerando contração das células musculares lisas)
é o principal estímulo para a ativação da NOS e síntese do NO. Entretanto,
a produção de NO pode ser dependente ou independente do aumento de Ca2+ citosólico.
Assim, a ativação da eNOS pelo Ca2+ se dá através do complexo calmodulina
ligada ao cálcio (CAM-Ca2+) e através de processos de fosforilação gerada
pela ação de fosfo-cinases específicas (PK).
Elevações
da concentração de Ca2+ citosólico, estimulam a formação do complexo
CAM-Ca2+ levando a ativação da eNOS. Além disso, diferentes proteínas interagem
com a eNOS e regulam sua atividade, como a proteína de estresse de 90kDa
(hsp90) que age como um modulador de ativação alostérica e promotor do (des)acoplamento
da eNOS. Caveolina-1 é uma proteína estrutural de caveolos (invaginações
de membranas plasmáticas), de grande interesse e tem demonstrado atenuar a
atividade da eNOS, levando à inibição da enzima em um processo reversível
modulado pela interação da CAM-Ca2+. O recrutamento de CAM-Ca2+ e hsp90 pode
deslocar a caveolina-1 levando a ativação da eNOS. Ativação de receptores
metabotrópicos, como os da bradicinina, provocam aumento do Ca2+ citosólico via
ativação da fosfolipase C, convertendo o PIP2 em inositol 1, 4, 5, trifosfato
(IP3) e diacilglicerol (DAG). A interação do IP3 com seu receptor, no retículo
sarcoplasmático, promove a liberação do Ca2+ para o citosol, levando à ativação
de canais iônicos (como os canais de Ca2+ dependentes de voltagem)
influenciados em sua maior parte pela PKC
Estímulos
que independem das concentrações de Ca2+ citosólico,
como a ativação de proteínas cinases, podem induzir a fosforilação da eNOS em
sítios de ativação, como resíduos de serina (Ser-1177, 635, 617, 615, 116) e
tirosina (Tyr-567) e em sítios de inibição como resíduos de tyrosina (Tyr-81) e
treonina (Thr-495). A fosforilação Ser-1177 é feita por um grande número de
proteínas cinases, incluindo a proteína cinase A (PKA), dependente da
concentração de AMP cíclico. Mecanismos de sinalização que culminam na
fosforilação da eNOS em Ser-1177 e Ser-615 foram inicialmente demonstrados em
estudos independentes que relataram a participação da via fosfatidil-inositol
3-cinase (PI3K)/AKT em resposta a estímulos mecânicos, como alterações do fluxo
sanguíneo (shear stress) ou receptores com ação catalítica, por exemplo,
a resposta endotelial à insulina. Eventos adicionais a estes estímulos
extracelulares, tais como alterações de ATP, hipóxia e fator de crescimento
endotelial vascular (VEGF) são descritos ainda para relacionar o papel da
proteína cinase ativada por 5’-AMP (AMPK) que; após a fosforilação da porção α
em Thr-172 e consequente ativação da subunidade catalítica, produz a
fosforilação da eNOS em Ser-1177. Mecanismos de transativação de proteínas
cinases dependentes da concentração de cálcio intracelular são descritas ainda
para explicar os mecanismos de ativação da eNOS. Foi demonstrado que a proteína
cinase dependente da cálcio-calmodulina (CaMKK) ativa cinases serina/treonina
específicas que agem sobre a proteína Ca2+-CaM, como a proteína cinase
dependente de cálcio calmodulina (CAMK) I, II e IV. Estudos com inibidores da
CAMK II demonstraram a ação desta proteína em fosforilar resíduos de Ser 615 e
Ser 1177 da eNOS, levando a sua ativação e produção de NO. Estímulos
decorrentes do estresse oxidativo levam à regulação de vias que causam a
ativação da proteína cinase C (PKC). A PKC interfere no processo de ativação da
eNOS por fosforilar o resíduo de Thr-495, uma vez que a desfosforilação desse
resíduo está associada ao aumento da atividade da eNOS e estímulos que elevam
as concentrações de Ca2+ intracelulares que culminam na ativação da PKC (Fig.
3 e 4).

Fig.
3: Fatores relaxantes derivados do endotélio. Representação da produção de
fatores dependentes da mobilização do cálcio na célula endotelial. PGI2:
Prostaciclina, NO: Óxido Nítrico, EDHF: Fator hiperpolarizante derivado do
endotélio (Adaptado de Vanhoutte, 1998)

Fig. 4: Representação de proteínas cinases envolvidas na fosforilação da eNOS. Alterações do fluxo sanguíneo produzem estimulação de mecanoreceptores sensíveis ao estiramento que induzem a fosforilação da eNOS por proteínas cinases AKT (Ser 1177, 617), AMPK (Ser 1177), PKA (Ser 1177, 635) e CAMKII (Ser 1177) (Adaptado de ZHANG et al. 2009).
2.2
REGULAÇÃO EXTRÍNSECA, se dá por fatores ou
mecanismos que começam fora do tecido ou órgão onde o vaso sanguíneo é
encontrado, agindo diretamente nos mecanismos de vasoconstrição ou
vasodilatação. É determinado principalmente pelo sistema nervoso autônomo
(fundamentalmente simpático), sistema vasopressinérgico e o sistema
renina-angiotensina. Em condições fisiológicas, a homeostase da pressão
arterial e da função circulatória são mantidas por uma complexa interação entre
esses 3 sistemas (Fig.5).

Fig
5. Interrelação fisiológica entre os sistemas adrenérgico,
vasopressinérgico e renina-angiotensina na homeostase da pressão arterial e
mecanismos selecionados de vasopressores farmacológicos. Receptor α1: alfa1-adrenérgico,
Receptor AT1R: angiotensina tipo 1, Receptor β1: beta1-adrenérgico,
Receptor β2: beta2-adrenérgico, Receptor V1: de
vasopressina 1. (extraída do artigo de Wieruszewski, P.M., Khanna, A.K.
Vasopressor Choice and Timing in Vasodilatory Shock. Crit Care 26, 76. Abril 2022)
Conforme o
Tratado de Fisiologia de Guyton[14],
o controle humoral do tônus vascular se realiza por mediadores vasoconstritores
e vasodilatadores
2.1.1
VASOCONSTRITORES
a)
Norepinefrina e Epinefrina. Mediadores sistema autônomo
simpático pertencente à classe das catecolaminas, secretadas internamente
(catecolaminas endógenas) ou administradas externamente (catecolaminas
sintéticas). As catecolaminas medeiam suas ações cardiovasculares predominantes
através dos receptores α1, β1, β2 e dopaminérgicos, cuja densidade e proporção nos
diferentes tecidos modulam as respostas fisiológicas e farmacológicas. A
epinefrina, é a principal catecolamina da glândula adrenal. Os efeitos de cada
uma dependerão do tipo de receptor catecolaminérgico (α ou β), do nível de
catecolaminas circulante (alto ou baixo) e da densidade de receptores em cada
tecido/órgão alvo, neste caso os receptores localizados nos vasos sanguíneos
(arteriais e venosos). A norepinefrina é uma substância vasoconstritora potente
já que tem feito predominantemente α. Já a epinefrina em doses baixas tem
efeito predominante β (vasodilatador β2 e inotrópico β1), mas efeito
predominante α em doses elevadas.
Os
receptores adrenérgicos ou adrenorreceptores pertencem à classe de receptores
ligados à proteína G e ão alvos das catecolaminas[15]. Existem
dois grupos principais de receptores adrenérgicos, α e β, apresentando vários
subtipos: os receptores α têm os subtipos α1 (um receptor acoplado a uma
proteína Gq) e α2 (um receptor acoplado Gi). Os receptores β possuem os
subtipos β1, β2 e β3. Todos os três estão ligados à proteína Gs, que por sua
vez estão ligadas à adenilciclase. Agonista obrigatório, assim, provoca um
aumento na concentração intracelular do segundo mensageiro AMPc. Na mesma direção,
os efetores do AMPc incluem proteína quinase dependente de AMPc (PKA), que
medeia alguns dos eventos intracelulares após a ligação do hormônio (Fig.6).
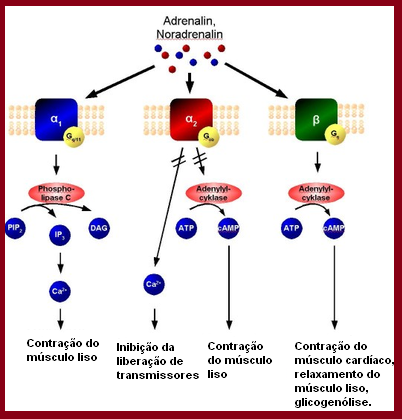
Fig 6. Receptores adrenérgicos α e
β (https://pt.wikipedia.org/wiki/Receptores_adren%C3%A9rgicos)
O
mecanismo dos receptores adrenérgicos. A adrenalina e noradrenalina são
os ligantes endógenos dos receptores, tanto do α1, α2 e β[16].
Receptores
α1, associados a proteína Gq, ativam a fosfolipase C, convertendo o
PIP2 em inositol 1, 4, 5, trifosfato (IP3) e diacilglicerol (DAG). A interação
do IP3 com seu receptor, no retículo sarcoplasmático, promove a liberação do
Ca2+ citosólico, resultando na contração do músculo liso. Receptores α2,
por outro lado, estão associados a proteína Gi, o que provoca uma diminuição da
atividade da adenil cilcase reduzindo a concentração de AMPc (normalmente AMPc
provoca contração de músculo liso), resultando no relaxamento do músculo liso (Fig.
7).

Fig.7 Mecanismo de ação dos
receptores α (Circulation. 2008;118:1047–1056)
Os
β receptores, estão associados à proteína Gs que aumenta a atividade da adenilciclase.
A função geral dos Gs é ativar as vias de sinalização intracelular em resposta
à ativação dos receptores acoplados à proteína G da superfície celular (GPCRs).
Os GPCRs funcionam como parte de um sistema de três componentes de
receptor-transdutor-efetor. O transdutor neste sistema é uma proteína G
heterotrimérica, composta por três subunidades: uma proteína Gα, como Gsα, e um
complexo de duas proteínas fortemente ligadas chamadas Gβ e Gγ em um complexo Gβγ.
Quando não estimulado por um receptor, Gα é ligado ao GDP e ao Gβγ para formar
o trímero inativo da proteína G. Quando o receptor se liga a um ligante
ativador fora da célula (como um hormônio ou neurotransmissor), o receptor
ativado atua como um fator de troca de nucleotídeo guanina para promover a
liberação de GDP e a ligação de GTP a Gα, o que leva à dissociação de Gα ligado
a GTP de Gβγ. Em particular, a Gsα ativada ligada a GTP liga -se à adenilil
ciclase para produzir o segundo mensageiro, o cAMP , que por sua vez ativa a
proteína cinase dependente de cAMP (também chamada de proteína cinase A ou PKA).Embora
cada Gsα ligado a GTP possa ativar apenas uma enzima adenilil ciclase, a
amplificação do sinal ocorre porque um receptor pode ativar múltiplas cópias de
Gs enquanto esse receptor permanece ligado ao seu agonista ativador, e cada Gsα
ligado a adenilil ciclase enzima pode gerar cAMP substancial para ativar muitas
cópias de PKA.
O
estímulo do receptor β1-adrenérgico, resulta em aumento da contratilidade
miocárdica como consequência da liberação do Ca2+ do reticulo sarcoplasmático
para o citoplasma e sua ligação ao complexo actina-miosina com troponina C. A
estimulação do receptor β2-adrenérgico presente nas células musculares
lisas vasculares resulta em ativação da fosfo cinase dependente de AMPc levando
a aumento da captação de Ca2+ pelo retículo sarcoplasmático e vasodilatação (Fig.
8).

Fig 8. Mecanismo de ação dos
receptores β (Circulation. 2008;118:1047–1056)
b)
Angiotensina II. Mediador do sistema renina-angiotensina-aldosterona (SRAA). A
angiotensina II é outra poderosa substância vasoconstritora. Apenas um milionésimo
de grama pode aumentar pressão arterial de uma pessoa em 50 mm Hg ou mais. A
angiotensina II (ATII) atua nos receptores da angiotensina I (ATI) encontrados
no endotélio das arteríolas em toda a circulação provocando vasoconstrição.
Essa sinalização ocorre por meio de uma proteína Gq, que ativa a fosfolipase C,
cascata da fosfatidil-inositol-bifosfato (PiP2) e, posteriormente, aumento do
cálcio citosólico e consequente vasoconstrição[17].
c)
Vasopressina. Mediador do sistema vasopressinérgico. A vasopressina também
chamada de hormônio antidiurético (HAD), é ainda mais poderosa que a
angiotensina II como vasoconstritor. É sintetizada nas células nervosas do hipotálamo
e transportada pelos axônios dos nervos para a hipófise posterior de onde é
finalmente secretada no sangue. O efeito vasoconstritor se dá através de
receptores V1 acoplados à proteína Gq, encontrados em alta densidade no músculo
liso vascular causando vasoconstrição pelo aumento do cálcio intracelular via
cascata da fosfolipase C, e fosfatidil-inositol-bifosfato (PiP2)[18].
Quando esses
sistemas são alterados por um insulto (por exemplo, sepse), o equilíbrio
homeostático é interrompido. O achado objetivo mais óbvio é a disfunção da
macrocirculação identificada pela medição direta da pressão arterial sistêmica,
embora os danos aos tecidos e à microcirculação ocorram regionalmente em
paralelo e até mesmo antes da evidência global de hipotensão[19].
No caso do choque vasoplégico séptico, embora um estado hiperdinâmico inicial induzido
pelo estresse frequentemente ocorra, a variabilidade total da frequência
cardíaca é reduzida, sugerindo comprometimento do sistema simpático[20].
Da
mesma forma, em situações de hipotensão, espera-se que a hipófise posterior secrete
as reservas endógenas de vasopressina, embora as concentrações plasmáticas
da vasopressina, no choque séptico vasoplégico tenham se mostrado
inadequadamente baixas (3,1 pg/ml) em comparação com outros estados de choque
como choque cardiogênico (22,7 pg/ml)[21]. Estudos
mostram que a concentração de vasopressina é elevada no choque séptico precoce,
mas diminui para a faixa normal na maioria dos pacientes entre 24 e 48 h, caso
o choque persista de forma contínua. Esse achado foi chamado de “insuficiência
relativa de vasopressina” pois, na presença de hipotensão, espera-se que os
níveis séricos de vasopressina aumentem. A importância desse achado
desconhecido[22] [23].
Finalmente,
apesar da ativação do sistema renina-angiotensina no choque, vários
receptores de angiotensina são regulados negativamente, contribuindo para a hiporreatividade
vascular, prejudicando também a secreção endógena de catecolaminas[24] [25].
2.2.2
VASODILATADORES
2.2.2.1
Bradicinina. Várias substâncias chamadas cininas causam uma poderosa
vasodilatação quando se formam no sangue e tecidos de alguns órgãos. As cininas
são pequenos polipeptídios que se formam a partir da ação de enzimas
proteolíticas sobre α2-globulinas no plasma ou fluidos teciduais. Uma enzima
proteolítica particular importância para este fim é a calicreína, que está presente
no sangue e fluidos teciduais na forma inativa. Esta calicreína inativa é
ativada por maceração do sangue, inflamação dos tecidos ou outros efeitos
químicos ou físicos semelhantes. À medida que a calicreína é ativada, ela atua imediatamente
sobre a α2-globulina para liberar um cinina chamada calidina, que é então
convertida por enzimas teciduais em bradicinina. Uma vez formada, a bradicinina,
esta persiste por apenas alguns minutos porque é inativada pela enzima
carboxipeptidase ou pela enzima conversora, a mesma enzima que também
desempenha um papel essencial na ativação da angiotensina I. A enzima ativada
calicreína é destruída por um inibidor de calicreína que também está presente
nos fluidos corporais. A bradicinina, é um peptídeo de 9 aminoácidos gerado em
condições fisiopatológicas como inflamação, trauma, queimaduras, choque e
alergia, e causa vasodilatação arteriolar poderosa e aumento da permeabilidade
capilar. Por exemplo, a injeção de 1 micrograma de bradicinina na artéria
braquial de uma pessoa aumenta o fluxo sanguíneo através do braço em até seis
vezes, e quantidades ainda menores injetadas localmente nos tecidos pode causar
um edema local marcado como resultado de um aumento do tamanho dos poros
capilares. A família de receptores de bradicinina é um grupo de receptores
acoplados à proteína G cujo principal ligante é a proteína bradicinina. Existem
dois receptores de bradicinina: o receptor B1 e o receptor B2. O B2, principal
receptor mediador das ações das cininas, é expresso em abundância pelas células
endoteliais vasculares e está presente na maioria dos tecidos, incluindo rins,
coração, músculo esquelético, SNC, ducto deferente, traqueia, intestinos, útero
e bexiga. Em geral, a distribuição e a ação dos B1 são semelhantes às dos B2. O
B1, por outro lado, é expresso em níveis baixos em condições normais, mas é
regulado positivamente em resposta a estímulos inflamatórios (por exemplo,
lipopolissacarídeo, endotoxinas e citocinas como IL-1β e TNF-α) e no cenário de
diabetes e lesão de isquemia-reperfusão. B2 liga tanto a bradicinina quanto a
calidina, enquanto a bradicinina quase não tem efeito no B1. Ambos se acoplam à
proteina Gq para ativar a fosfolipase C (PLC), o que resulta em hidrólise de
fosfoinositídeo, produção de DAG e mobilização de Ca2+ para o citosol (estímulo
que ativa a NOS e a síntese de NO). Eles também podem atuar através de Gi para
inibir a adenilciclase. Além disso, o receptor estimula as vias da proteína
quinase ativada por mitógeno. Os efeitos fisiológicos da ativação do receptor
de bradicinina são mediados pela geração de NO derivado da sintase de NO
endotelial e prostaglandinas. A ativação de B2 leva a um aumento nas
concentrações de cálcio intracelular nas células endoteliais vasculares. No
entanto, a vasodilatação induzida pela bradicinina não é abolida pela
coadministração de NO sintase e inibidores de COX, o que indica que efetores
adicionais provavelmente também estão envolvidos, possivelmente um fator
hiperpolarizante derivado do endotélio (EDHF). Além disso, através da ligação a
B1 e B2, bradicinina também aumenta a expressão de NO sintase induzível (iNOS),
pelo menos em roedores. É muito difícil induzir o gene iNOS em tecidos humanos,
especialmente no endotélio vascular. Todos esses mecanismos levam a vasodilatação
arteriolar[26] [27].
2.2.2.2
Histamina. A histamina é liberada em quase todos os tecidos do corpo se o
tecido ficar danificado ou inflamado ou se for exposto a uma reação alérgica. A
maior parte da histamina deriva dos mastócitos em tecidos danificados e de basófilos
sanguíneos. A histamina tem um poderoso efeito vasodilatador nas arteríolas e,
como a bradicinina, tem a capacidade de aumentar muito a porosidade capilar,
permitindo o vazamento de fluido e proteínas plasmáticas aos tecidos em muitas
condições patologias, induzindo edema. Os receptores de histamina são
receptores transmembrana. A histamina pode ser liberada como transmissor em
preparações neuronais ou como mediador de uma resposta inflamatória pelos
mastócitos. Atualmente, quatro receptores de histamina foram identificados. A
histamina é um potente mediador em muitos processos fisiológicos, causando
vasodilatação ou vasoconstrição, estímulo da frequência cardíaca e a
contratilidade e a contração dos músculos lisos do intestino e das vias aéreas,
dependendo do tipo de receptor estimulado. O receptor H1 da histamina, é
expresso em vários tecidos, incluindo o cérebro, músculo liso da vasculatura
e vias aéreas. O receptor H2 da histamina, é ubiquamente expresso em vários
tecidos, incluindo o estômago, coração e cérebro. O receptor H3 da histamina, é
expresso principalmente nos neurônios, como auto e heterorreceptor
pré-sináptico. O receptor de histamina H4, é um receptor quimiotático, expresso
principalmente em eosinófilos, mas também em mastócitos, células dendríticas e
células T. Também são expressos no epitélio do trato gastrointestinal. Os
receptores H1 acoplam-se a Gq que regula a mobilização de Ca2+ para o citosol
(estimulando a NOS e a produção de NO), os receptores H2 acoplam-se a Gs
para estimular o AMP cíclico e os receptores H3 e H4 ambos acoplam a Gi/o para
inibir o acúmulo de AMP cíclico. Os receptores de histamina estão amplamente
distribuídos no corpo, com um ou mais receptores expressos em níveis
significativos em neuronal, músculo liso (vascular, gástrico e brônquico),
sangue, coração, sistema imunológico e sistema endócrino[28] [29]. Os
receptores H1 e H2 que são os que mediam a vasodilatação. Os receptores H2
estão localizados principalmente nas células musculares lisas dos vasos
sanguíneos e os efeitos vasodilatadores são mediados pelo AMPc. Os receptores
H1 residem principalmente nas células endoteliais, e sua estimulação leva à formação
de NO[30].
2.2.2.3
CONTROLE DO TÒNUS VASCULAR POR ÍONS E OUTROS FATORES QUÍMICOS
Sabe-se
que o mecanismo de contração/relaxamento do músculo liso vascular e do músculo
cardíaco depende da ocorrência previa do fenômeno elétrico (despolarização e
repolarização). Por sua vez, o fenômeno elétrico depende do movimento de íons
(gradiente químico e elétrico) entre o espaço extracelular e intracelular que
ocorre tanto no tecido especializado nervoso (fibras do sistema nervoso, do
sistema autonômico cardíaco e do sistema de condução) quanto no tecido muscular
liso dos vasos e muscular cardíaco. Os principais íons determinantes do
fenômeno elétrico são o sódio (Na⁺), o potássio (K⁺) e o cálcio (Ca⁺⁺). A
concentração fisiológica ou patológica destes íons nos compartimentos intra e
extracelular determinará o estado de polarização normal (potencial de repouso
normal negativo), hiperpolarização (redução do potencial de repouso para
valores mais negativos) ou de despolarização (aumento do potencial de repouso
tornando-o menos negativo até valores positivos). Há que se levar em
consideração que a concentração desses íons nos seus respectivos
compartimentos, e sua movimentação através dos seus respectivos canais se dá através
de gradientes químico (gradiente de concentração) e elétrico (polaridade do
compartimento atrai ou repele o íon com carga oposta ou igual respectivamente).
Importante lembrar também que a abertura de canais iônicos (de Na⁺, K⁺ e Ca⁺⁺)
muitas vezes depende de atingir uma determinada voltagem (diferença de
potencial em mV) entre os compartimentos.
a)
Calcio. Um aumento na concentração de íons de cálcio extracelular,
favorece o gradiente para o intracelular (cálcio normalmente está em maior
concentração no extracelular) causando vasoconstrição devido ao efeito do
cálcio em estimular a contração do músculo liso vascular. O meio extracelular
sem cálcio impede a sua internalização tanto por via membrana celular como via
retículo sarcoplasmático, evitando que haja contração dos miofilamentos. Mesmo
havendo potencial de ação, não há contração muscular.
b)
Potássio. A hipocalemia hiperpolariza o potencial de membrana ou repouso
(favorece o gradiente intra – extracelular de potássio, fazendo intracelular e
o potencial de repouso mais negativos), reduzindo, assim, também a excitabilidade.
A hipocalemia torna o potencial de repouso mais negativo (hiperpolarização) embora
numericamente o potencial se reduza. Ao se afastar mais do potencial de ação a
excitabilidade diminui. A hipercalemia faz com que o potencial de membrana (de
repouso) se torne menos negativo. Se esse valor ainda tiver abaixo do potencial
de ação, tornará a celular muscular mais excitável, mas se estiver acima do
potencial de ação diminuirá a excitabilidade pela inativação dos canais rápidos
de Na+ (são voltagem dependentes e precisa que o potencial de repouso normal
seja atingido para se abrirem)[31].

Fig. 9. Efeitos do potássio no
potencial de ação (ref. 31).
Na
hipercalemia, aumento na concentração de íons potássio no extracelular, retarda
o período refratário na fase de repolarização (saída de potássio do intra para
o extracelular) fazendo com que a célula muscular já relaxada (pelo fechamento
dos canais de cálcio) se torne refratária a nova despolarização (e nova
contração). Assim hipercalemia favorece o relaxamento do musculo liso vascular
e do próprio miocárdio podendo gerar parada cardíaca em diástole. Estudos de
cardioplegia hipercalcêmica permitiram observar que o potássio extracelular
elevado (10-40mM) altera o potencial de repouso dos miócitos de -85mV para uma
faixa que varia de -65 mV a - 40mV, inativando os canais rápidos de sódio. Esse
novo potencial de repouso bloqueia a condução do potencial de ação miocárdico,
induzindo assim a parada cardíaca despolarizada (em diástole). Entretanto, ela
não inativa completamente os canais lentos de sódio (janela do sódio),
aumentando sua concentração intracelular de maneira lenta. Associado a isso, o
canal de cálcio tipo L (dihidropiridínico), que é ativado com potencial entre
-20mV a -30mV, faz com que o cálcio adentre o citosol, fenômeno esse chamado de
janela do cálcio. A seguir, a bomba antiporte Na+/Ca2 + é ativada ao contrário,
retirando sódio do intracelular e internalizando o íon cálcio. Esse panorama
intracelular de cálcio elevado pode levar a contração do miócito mesmo sem
deflagrar potencial de ação[32]. Na
hipocalemia, ocorre o fenômeno inverso a excessiva eletronegatividade externa
durante a despolarização (redução de sódio com menos potássio extracelular), retarda
o fechamento dos canais de cálcio perpetuando a contração do musculo liso
vascular (vasoconstrição) e cardíaco, podendo gerar parada cardíaca em sístole.
c)
Magnésio. Um aumento na concentração de íons de magnésio causa
vasodilatação poderosa porque os íons de magnésio inibem a contração do músculo
liso.
d)
Acidose/alcalose. Um aumento na concentração de íons hidrogênio (diminuição do pH) promove
dilatação das arteríolas. Ao contrário, uma ligeira diminuição na concentração
de íons hidrogênio causa constrição arteriolar.
e)
Ânions que têm efeitos significativos sobre vasos sanguíneos são acetato
e citrato, que causam graus leves de vasodilatação.
F)
CO2 e O2. Um aumento na concentração de dióxido de carbono (hipercapnia) pode
levar a acidemia grave (pH < 7,2) contribuindo para vasoconstrição
arteriolar pulmonar, vasodilatação arteriolar sistêmica moderada na maioria dos
tecidos (porém marcada no cérebro). Além disso, o dióxido de carbono no sangue,
que atua no centro vasomotor cérebro, tem um efeito extremamente indireto poderoso,
transmitido através do sistema vasoconstritor do nervo simpático, que causa uma
vasoconstrição generalizada em todo o corpo.
A hipóxia aguda causa vasoconstrição pulmonar com aumento da RVP. A hipóxia aguda é associada a um
aumento, e não a uma diminuição da produção de NO, provavelmente pelo “shear
stress” causado pela vasoconstrição hipóxica, que provavelmente envolve a
formação de espécies reativas de oxigênio, endotelina-1 ou produtos do
metabolismo do ácido araquidônico. A hipoxia resultante da oferta reduzida de
oxigênio ou do aumento da demanda de oxigênio causa, no entanto, vasodilatação
sistêmica. A vasodilatação sistêmica induzida pela hipóxia pode ser direta (O2
inadequado para sustentar a contração do músculo liso) ou indireta pela produção
de metabólitos vasodilatadores[33].
III.
CAUSAS E FISIOPATOLOGIA DO CHOQUE VASOPLÉGICO[34]
Vasoplegia, é
um estado caraterizado por resistência vascular sistêmica (RVS) anormalmente
baixa, que se manifesta como hipotensão grave na presença de um débito cardíaco
normal ou aumentado. A RVS é definida como a razão entre a pressão de perfusão tecidual
e o débito cardíaco (PAM − PAD) /DC).
Entre
as principais causas se descrevem:
1.
Sepse. A causa mais comum de vasoplegia em cuidados intensivos é a sepse.
A incidência depende da definição utilizada e da população de pacientes analisada.
A
sepse resulta de uma complexa interação entre o microrganismo infectante e a
resposta imune, a partir de células fagocíticas, criando um estado
fisiopatológico pró-inflamatório e pró-coagulante no hospedeiro. A resposta do
hospedeiro inicia-se quando células da imunidade inata, particularmente
macrófagos, reconhecem e se ligam a componentes de agentes microbianos
patógenos, desencadeando diversas reações e rotas metabólicas que resultam na
produção e na liberação de citocinas pró-inflamatórias, com consequente lesão
celular. Estabelece-se a falência dos órgãos a partir da disfunção
mitocondrial, havendo aumento do déficit de oxigênio pelos tecidos. Essa ligação das células da
imunidade inata se dá através de receptores de reconhecimento padrão localizados
nas suas membranas, que incluem os Toll-Like Receptors (TLRs), CD14, componentes
da via alternativa do complemento, lectina ligadora de manose. Os dois
primeiros são os de maior relevância.
A
família dos receptores Toll-Like (TLRs) é a mais importante dos sistemas de
receptores de reconhecimento padrão associado a patógenos nos seres humanos. Os
TLRs são receptores transmembrana que detectam endotoxina e numerosos
mediadores microbianos diferentes, incluindo componentes bacterianos, fúngicos,
virais e parasíticos. Os TLR-2 reconhecem os peptideoglicanos das bactérias
gram-positivas, enquanto os TRL4 reconhecem os lipopolissacarídeos (LPS) das
gram-negativas. Os TLRs desencadeiam vários eventos intracelulares que resultam
na translocação do NF-kB para o núcleo celular, um fator de transcrição que
promove a expressão gênica de moléculas pró-inflamatórias, como fator de
necrose tumoral alfa (TNF-α) e a interleucina-1 beta (IL-1β) assim como também
citocinas anti-inflamatórias, como interleucina-10 (IL-10). Juntos, TNF-α e
IL-1β, ampliam a imunidade inata por ativarem a imunidade adaptativa (Fig.
10). Dessa forma, a partir da ativação das células B, elas liberam
imunoglobulinas facilitadoras da apresentação de antígenos para as células
fagocitárias. O feedback positivo é feito pelas células T helper tipo 1 (Th1),
secretando citocinas pró-inflamatórias (TNF-α e IL-1β). Em oposição a isso,
atuam as células T helper tipo 2 (Th2), secretando interleucinas
anti-inflamatórias (IL-4, IL-10). Grandes quantidades de citocinas
pró-inflamatórias na corrente sanguínea contribuem para a progressão do
processo séptico. Tanto o TNF-α quanto a IL-1β podem causar febre, hipotensão,
leucocitose, indução de outras citocinas pró-inflamatórias e ativação
simultânea da coagulação e fibrinólise. Os efeitos das citocinas
pró-inflamatórias determinam o aumento e a expressão das moléculas de adesão
dos leucócitos, que, quando ativados, migram da corrente sanguínea para os
tecidos inflamados através das células endoteliais. Os neutrófilos ativados
também causam aumento da permeabilidade vascular, causando edema tecidual. Por
fim, o óxido nítrico (NO), um potente vasodilatador liberado pelas células
endoteliais, passa a apresentar papel fundamental na patogênese do choque
(choque séptico). A ativação dos monócitos e dos macrófagos e a intensa ação
dos mediadores iniciais acarretam a síntese de outras citocinas, como IL-6,
IL-8, IL-10 e HMGB1 (high mobility group protein box 1), com vários efeitos
sinérgicos e antagônicos na resposta inflamatória. A IL-6, principalmente, tem
papel relevante, pois desencadeia a reprogramação da expressão gênica hepática,
a denominada resposta de fase aguda. A resposta de fase aguda também induz a
formação de várias proteínas que limitam a inflamação, entre elas, a proteína
C-reativa (PCR).

Fig.
10. Toll-like receptor 4 (TLR4): Local para novas intervenções (novas drogas)
com possibilidade de bloquear a transdução de sinal do receptor, o qual, na
sequência, inibe a liberação de citocinas infl amatórias, IL-1 e TNF, e suprime
o desenvolvimento da sepse grave (Ref. 35)
O
CD14 (cluster of differentiation 14) é um gene humano que codifica uma
proteina componente do sistema imune inato. Trata-se de uma proteína de 53-55kD
que não possui porção citoplasmática e atua como um receptor de uma ampla gama
de microrganismos onde reconhece diferentes estruturas de lipopolissacarídeos
(LPS). O CD14 pode ser encontrado sob duas formas: na superfície de monócitos,
macrófagos e neutrófilos (mCD14 ou CD14 de membrana) ou na forma de CD14
solúvel (sCD14). Tanto o mCD14 quanto o sCD14 desempenham um importante papel
na geração de uma resposta imune inata contra patógenos bacterianos. A ativação
do sistema imune inato por componentes bacterianos pode ser modulada pela
diferente expressão do mCD14 e variações do sCD14.[35] [36] [37] [38]
Como
mencionado acima, o fator de transcrição mais importante na sepse é o NFκB
desencadeado pelos TLRs. NF-kB é um fator nuclear (NF) que, uma vez ativado por
agentes como lipopolissacarídeos, possui a capacidade de ligar-se a uma
sequência de 10 pares de bases na região promotora do gene que codifica a
cadeia leve k das moléculas de anticorpo das células B (kB). A ativação de NFκB
ocorre pelo reconhecimento de moléculas de superfície típicas de bactérias
gram-positivas e gram-negativas através da via de sinalização mediada pelo
receptor Toll-like (TLR). Além disso, o NFκB é desencadeado por citocinas
pró-inflamatórias, como TNF-α e IL-1β, através de um mecanismo de feedback
positivo estabelecido à medida que citocinas pró-inflamatórias são liberadas
por células imunes habilitadas (ou seja, ativadas por NFκB).
Os
interferons são outro grupo de citocinas afetam diversas funções celulares,
incluindo respostas antivirais, antimicrobianas, de apoptose e de controle do
ciclo celular. Esta família de citocinas é dividida em três classes: tipo I (α,
β, ω e т), tipo II (γ) e tipo III (λ). O interferon-γ (IFN), único da classe
II, é uma citocina produzida principalmente por células do sistema imune ativadas
para combater infecções virais e bacterianas. Sua expressão pode ativar o NF-κB,
bem como também pode ser induzida em quase todos os tipos celulares pela
transcrição de genes mediada pelo NF-κB. Esta citocina exerce seus efeitos
através da ligação com seu receptor, o IFNγR1 ou IFNγR2. Células musculares lisas
vasculares primárias respondem à estimulação com IFN e TNF-α produzindo
concentrações significativas de NO, evidenciando que estas células além de
serem alvo do NO produzido pelas células endoteliais, são também fontes
geradoras de NO. As concentrações elevadas de NO, bem como a atividade
aumentada do GMPc nos vasos em resposta a endotoxinas e citocinas, estão
relacionados com a hipotensão e hiporeatividade a vasoconstritores durante o
choque séptico[39].
Além
de citocinas, o NFκB induz o aumento da expressão de quimiocinas, moléculas de
adesão e enzimas como a oxido nítrico sintetase induzida (iNOS) e a ciclooxigenase-2
(COX-2) através da regulação da expressão gênica. Induz, portanto, A influência
do NFκB na liberação de TNF-α e na expressão de iNOS e COX-2 é citada como uma
das razões da hipotensão sistêmica secundária à vasodilatação, diminuição da
responsividade vascular aos vasoconstritores e redução da contratilidade
cardíaca. Nesse contexto, supõe-se que a inibição do NFκB reduz a disfunção
cardíaca. Em camundongos estimulados com lipopolissacarídeo (LPS), o inibidor
específico de NFκB SUN C8079 mostrou resultar em diminuição da expressão gênica
de TNF-α e iNOS, e essa diminuição foi acompanhada por uma redução
dose-dependente na mortalidade. Resultados semelhantes foram obtidos com outro
inibidor específico de NFκB, IRFI-042, que reduziu a liberação de TNF-α e a
letalidade em ratos tratados com endotoxina. O antioxidante ditiocarbamato de
pirrolidina, outro inibidor seletivo, atenua de forma dose-dependente os complexos
NFκB/DNA e prejudica a diminuição da PAM induzida por LPS em ratos. No entanto,
a inibição do NFκB também tem alguns efeitos prejudiciais. Em um estudo,
camundongos sem uma subunidade NFκB p50, foram incapazes de se defender com
sucesso contra a infecção por Streptococcus pneumoniae. O uso de inibidores de
NFκB como estratégia terapêutica é, portanto, limitado devido à interação com
sua função de defesa do hospedeiro, importante para eliminar patógenos
causadores de sepse. A indução de iNOS mediada por sepse leva ao aumento da
produção de NO. O próprio NO ativa a guanilato ciclase solúvel, que aumenta o
monofosfato de guanosina cíclico (cGMP) e, assim, desencadeia o relaxamento do
músculo liso miocárdico e vascular. O tratamento de camundongos com LPS resulta
na expressão de mRNA de iNOS, que é acompanhada por contração reduzida dos
anéis carotídeos em comparação com camundongos não sépticos e camundongos
deficientes em iNOS. Da mesma forma, a reatividade do músculo liso aórtico à
catecolamina norepinefrina está diminuída em camundongos sépticos. Corações de
ratos tratados com LPS demonstraram desenvolver níveis aumentados de iNOS e NO
com diminuição do trabalho e da eficiência cardíaca. Em humanos, a capacidade
de resposta às catecolaminas em corações com e sem falha pré-tratados com LPS é
reduzida, enquanto o mRNA de iNOS é altamente expresso em todas as preparações,
mas sem cGMP aumentado. Assim, a liberação de mediadores endógenos, como óxido
nítrico (NO), bem como citocinas pró-inflamatórias, como fator de necrose
tumoral alfa (TNF-α) e interleucina-1 beta (IL-1β) tem sido associada à
disfunção cardíaca e vascular. Depressão miocárdica central direta e
insuficiência regional de células musculares lisas vasculares com
vasoconstrição ineficaz foram descritas. No entanto, estratégias para resolver
desequilíbrios de mediadores usando inibidores de NO, TNF-α, IL-1β ou
prostaglandinas/prostaciclinas não melhoraram a sobrevida de pacientes com
sepse. Diferentes abordagens terapêuticas para a produção de iNOS e NO foram
avaliadas. Em modelos de sepse baseados em LPS de ratos e coelhos, o tratamento
com um inibidor específico de iNOS (1400W) reduziu o nível sanguíneo de NO e a
hipotensão em ratos, mas não em coelhos. Na sepse tardia, mas não precoce,
1400W melhorou a contração cardíaca em ratos. O aumento do trabalho cardíaco e
da função contrátil também foi demonstrado em associação com os inibidores de
iNOS mercaptoetil guanidina, aminoguanidina ou azul de metileno em modelos de
ratos e camundongos. Pacientes humanos com sepse que receberam azul de metileno
desenvolveram aumento da PAM e da resistência vascular sistêmica, enquanto as
alterações no índice cardíaco foram dose-dependentes e ocorreram eventos
adversos como metemoglobinemia, anemia hemolítica e alterações na função
pulmonar. No geral, não há vantagem na sobrevida. O tratamento de pacientes
humanos com sepse usando inibidores não seletivos da NOS (análogos competitivos
da L-arginina) produziu resultados benéficos e prejudiciais. O inibidor não
seletivo de NOS L-NMMA supostamente previne uma diminuição induzida por LPS na
contratilidade cardíaca e na função ventricular esquerda. Outro inibidor não
seletivo da NOS, 546C88, foi associado a maior mortalidade (59%) em comparação
com placebo (49%) no dia 28 em pacientes com sepse e um aumento do número de
eventos adversos, como insuficiência cardíaca, débito cardíaco diminuído e
hipertensão pulmonar ocorreu com o uso deste inibidor. Coletivamente, os
achados dos estudos diferem amplamente entre as diferentes espécies animais,
bem como entre os modelos animais e humanos; isso é problemático em termos de
desenvolvimento de novas terapias. Além disso, o sucesso terapêutico da
inibição da iNOS pode depender da dose, tempo de administração e fase da sepse.
Mais estudos são claramente necessários para avaliar os potenciais benefícios
dos inibidores de iNOS para pacientes com sepse. Outras opções terapêuticas que
foram avaliadas para a sepse incluem a inibição de mediadores de condução da
inflamação, como prostaglandinas (por exemplo, PG2), prostaciclinas (por
exemplo, PGI2), TNF-α e IL-1β. No entanto, nenhuma melhora contundente na
sobrevida de pacientes humanos com sepse foi encontrada com base em tais
tratamentos. Ensaios multicêntricos randomizados mostraram apenas uma pequena
redução de mortalidade de 3,5% quando vários anticorpos anti-TNF-α são
administrados a pacientes com sepse. Portanto, essas abordagens não têm
relevância prática no atendimento médico diário de pacientes com sepse[40].
2.
Cirurgia cardíaca. Vasoplegia e choque vasoplégico que ocorrem em pacientes após
cirurgia cardíaca são a segunda causa mais comum. O diagnóstico é mais
complexo, pois existe um risco sempre presente de débito cardíaco prejudicado
contribuindo para a hipotensão, cuja causa deve ser determinada precocemente,
diferenciando entre pré-carga reduzida por sangramento, contratilidade
miocárdica prejudicada ou ocorrência de tamponamento cardíaco. O segundo
principal fator complicador é o uso frequente de inotrópicos vasodilatadores que
influenciam diretamente o tônus vascular. Portanto, embora não exista uma
definição consensual, existem várias definições de trabalho que combinam i)
hipotensão na ausência de um estado de baixo débito cardíaco e ii) ausência de
infecção; critérios adicionais também podem incluir a ausência de inotrópicos
vasodilatadores, como dobutamina ou milrinona, ou presença de evidência de
hipoperfusão tecidual e vários regimes de tratamento considerados, incluindo o
uso de vasoconstritores alternativos. A síndrome vasoplégica é comum após
procedimentos cardiovasculares e está associada a piores desfechos,
principalmente pelas alterações de perfusão, como injúria renal aguda, duração
da internação hospitalar e morte. A taxa de ocorrência varia de 5 a 25% no
pós-operatório de pacientes sem fatores de risco conhecidos, mas naqueles com
predisposição conhecida para a síndrome a prevalência pode variar de 30 a 50%
dos casos. Dentre os fatores conhecidos como predisponentes para a vasoplegia
pós-operatória estão incluídos o uso de inibidores da enzima conversora da
angiotensina (iECA), betabloqueadores e comorbidades prévias de impacto
significativo, disfunção sistólica pré-operatória e, no intraoperatório,
necessidade de altas doses de vasopressores antes ou durante a circulação
extracorpórea (CEC), alta temperatura durante o bypass e CEC prolongada. A CEC
causa uma resposta inflamatória profunda que resulta na produção anormal de
óxido nítrico, depleção do ATP e acidemia aumentada no músculo liso vascular,
resultando em uma diminuição da ativação de proteínas contráteis, levando,
assim, à vasodilatação. Ao mesmo tempo, o armazenamento endógeno de
vasopressina é rapidamente consumido, compondo o efeito vasodilatador da
síndrome e posterior choque vasoplégico[41] [42] [43].
3.
Cirurgia não cardíaca. Hipotensão devido à vasodilatação
em pacientes após cirurgia não cardíaca de grande porte geralmente se manifesta
como necessidade de vasopressores para manter uma PAM adequada após
ressuscitação apropriada para restaurar a euvolemia, e sua incidência raramente
é relatada. Os fatores de risco relatados incluem cirurgia prolongada e
necessidade significativa de transfusão de sangue. Onde a internação
pós-operatória em um ambiente de cuidados intensivos é rotina, o uso de
vasopressores no período pós-operatório para manter a pressão arterial após a
otimização do estado hídrico é comum. Embora os vasopressores possam ser
necessários para neutralizar os efeitos vasodilatadores sistêmicos do bloqueio
neuroaxial, como a analgesia epidural, onde as necessidades são significativas
em um paciente adequadamente ressuscitado, isso deve ser considerado
vasoplegia.
4.
Queimaduras, traumas e pancreatite. São condições unidas por lesão
tecidual significativa, com consequente hipermetabolismo, inflamação sistêmica
e predisposição ao desenvolvimento de disfunção orgânica. A vasoplegia pode ser
considerada uma dessas disfunções orgânicas e é uma complicação reconhecida de
politraumatismos, queimaduras e, mesmo na ausência de infecção, pancreatite
grave – onde a vasoplegia está associada a resultados adversos.
Fisiopatologia
Como
visto anteriormente a resistência vascular sistêmica (RVS) normal é determinada
pelo diâmetro arteriolar, regulado pela atividade contrátil das células
musculares lisas vasculares (CMLV) da túnica média das arteríolas, cujo tônus é
regulado pela concentração de cálcio intracelular (Ca2+). A contração da CMLV é
impulsionada por um aumento na concentração de Ca2+ citosólico através da
liberação de Ca2+ armazenado do retículo sarcoplasmático, bem como influxo de
Ca2+ do extracelular para o intracelular através de canais de Ca2+ dependentes
de voltagem. O relaxamento das CMLV é impulsionado por uma queda no Ca2+
citosólico, devido à captação de Ca 2+ pelo retículo sarcoplasmático e expulsão
de potássio (K+) ou de Ca2+ (via canais de K+ e bombas de Ca2+ATPase) para o
espaço extracelular, resultando em hiperpolarização e vasodilatação celular. O
tônus vascular é, portanto, dependente da relação influxo/efluxo de Ca2+, que
por sua vez é regulada por mecanismos intrínsecos e extrínsecos.
Entre
os mecanismos intrínsecos, temos o oxido nítrico (NO) que se difunde
livremente do interior do endotélio onde é produzido para as CMLV vizinhas e para
a corrente sanguínea causando vasodilatação, inibição da proliferação de CMLV,
ativação plaquetária e adesão leucocitária. Os autacoides inflamatórios,
incluindo bradicinina e trombina, aumentam a produção de NO e a vasodilatação
pela ativação da eNOS. Além disso, citocinas inflamatórias e estímulos
bacterianos como o lipopolissacarídeo (LPS) das bactérias gram negativas e
os peptidoglicanos (PG) das gram positivas, induzem a ativação da terceira
isoforma de NOS independente de cálcio (iNOS). Isso resulta em um aumento do NO
de duas a três vezes em magnitude acima da linha de base e é um dos principais
fatores da vasodilatação aguda no choque. A administração de inibidores não
seletivos da NOS mostrou estar associada à melhora da hemodinâmica em pacientes
com choque séptico, mas também a aumento da mortalidade, provavelmente pelo
impacto da inibição da NOS nas células imunes e na produção cardíaca de NO. As
terapias que visam a vasculatura e modulam, mas não bloqueiam totalmente a
síntese de NO podem oferecer um perfil mais favorável àquelas previamente
testadas até o momento em ensaios clínicos. Dentre os prostanóides, a
prostaciclina (PGI2) produzida pelo endotélio vascular causa agregação
plaquetária e induz vasodilatação. A produção de prostaciclina é grandemente
aumentada na inflamação e contribui para a vasodilatação. Uma ampla gama de citocinas
incluindo interleucina 1 (IL-1), fator de necrose tumoral α (TNF-α), hipóxia e
LPS, provocam a indução da isoforma COX-2 e aumento da síntese de PGI2 mediada pela
prostaciclina sintase (PGIS), que leva a vasoplegia. Ensaios terapêuticos de
inibição não seletiva de COX na sepse mostraram-se inconclusivos, com quaisquer
efeitos benéficos sobre o grau de vasoplegia mediada por PGI2 provavelmente
compensados por outras ações mediadas por prostaglandinas. Um prostanóide de
curta duração, o tromboxano A2 (TXA2) se opõe às ações do PGI2 e promove
vasoconstrição e agregação plaquetária. Portanto, o TXA2 foi implicado como um
potencial fator causador no aumento do risco de isquemia cardíaca em pacientes
tomando inibidores de COX2. O TXA2 regula o tônus vascular através da ligação
aos receptores de tromboxano-prostanóide (TP) no músculo liso vascular e, de
acordo com outros agentes, promove influxo de cálcio e vasoconstrição. A endotelina
1 (ET1) atua como vasoconstritor ao ativar os receptores de endotelina A
(ETA) nas CMLV, que conduzem a elevação do Ca2+ intracelular e a vasoconstrição.
Subtipos de receptores de endotelina B (ETB), encontrados no endotélio e no
músculo liso vascular, atuam como um mecanismo autorregulador para controlar o
tônus basal por meio da vasodilatação e contração do músculo liso. Em
condições de estresse inflamatório, no entanto, ET1 tem efeitos potencialmente
deletérios através da ativação de uma série de vias de sinalização, aumentando
a síntese de IL-1, TNF-α e IL-6. O bloqueio seletivo e não seletivo dos
subtipos de receptores ET mostrou-se promissor em uma variedade de modelos
animais. Espécies reativas de oxigênio (ROS) podem aumentar devido ao desacoplamento
das enzimas NOS endoteliais levando a disfunção mitocondrial. O ânion
superóxido pode reduzir o NO para formar peroxinitrito (ONOO−), que atua como
um poderoso agente oxidante que provoca disfunção celular e vasoplegia. Sob
condições fisiológicas, o ânion radical superóxido é metabolizado pela
superóxido dismutase (SOD). Os mecanismos não enzimáticos para o metabolismo do
superóxido são mediados pelo ácido ascórbico e pelo ácido úrico. Em estados de
choque, o excesso de produção de NO resulta em excesso de produção de ONOO−,
que pode ser atenuado por antioxidantes. ROS também podem causar a desativação
de catecolaminas, um fenômeno que pode ser revertido pela administração de um análogo
sintético da superóxido dismutase. O sulfureto de hidrogénio (H2S) é
sintetizado a partir do aminoácido L-cisteína mediado pela
cistationina-β-sintase ou cistationina-γ-liase dependente de vitamina B6. O H2S
difunde-se prontamente no músculo liso vascular e em baixas concentrações pode
ter efeitos citoprotetores, embora na sepse as concentrações sejam significativamente
elevadas. Em concentrações mais altas, o H2S contribui para o desenvolvimento
do choque vasoplégico através de uma série de ações dependentes de oxigênio,
incluindo a inibição do citocromo C oxidase com comprometimento da função
mitocondrial, ativação dos canais de potássio dependentes de ATP e inibição da
atividade da enzima conversora de angiotensina endotelial. Além disso, o H2S
interage com o NO, o que pode atenuar seus efeitos. H2S também foi sugerido
como um potencial agente terapêutico que leva ao desenvolvimento de um estado
semelhante à hibernação citoprotetora. Os animais tratados com H2S são
protegidos tanto da hipóxia letal como da hemorragia. Esta descoberta levou ao
estudo pré-clínico do tratamento com H2S na modulação dos efeitos deletérios da
lesão de isquemia-reperfusão em modelos experimentais, incluindo lesão
miocárdica porcina. A hiperpolarização celular associada a efluxo de
potássio através dos canais de potássio dependentes de ATP, é um mecanismo
importante para a regulação do potencial de membrana das CMLV. A hiperativação
dos canais de potássio resulta na hiperpolarização da célula, com a resultante
inativação dos canais de cálcio dependentes de voltagem, levando a vasodilatação.
Além dos mediadores derivados do endotélio, vários fatores circulantes podem
conduzir a disfunção vascular mediada pelos canais de potássio, incluindo
hipóxia, pH reduzido e aumento do lactato circulante. A disfunção vascular
induzida por estresse inflamatório como a endotoxina levou à hipótese de que a
inibição dos canais de potássio pode oferecer uma nova estratégia terapêutica.
Modelos animais mostraram melhorias hemodinâmicas após a inibição com glibenclamida,
um bloqueador específico do canal de potássio sensível ao ATP. No entanto, ensaios
controlados randomizados de fase 2 em seres humanos não demonstraram benefício,
e as preocupações com os efeitos não vasculares limitam sua utilidade
potencial.
Entre
os mecanismos extrínsecos, temos a resistência às catecolaminas, levando
a perda da sua eficácia vasoconstritora. Modelos animais sugerem que em
estágios mais avançados da sepse, a expressão do adrenoceptor alfa-1 cai,
resultando em resistência periférica à norepinefrina. Em estudos humanos, a
expressão de receptores periféricos parece estar relacionada à gravidade da
doença, com expressão aumentada na doença leve e expressão reduzida observada
na sepse grave, sugerindo que em pacientes com vasoplegia pode ocorrer um
padrão semelhante ao observado em modelos de roedores. A insuficiência adrenal
associada o choque é um mecanismo também bastante estudado. Os
glicocorticóides conduzem diversas respostas teciduais na inflamação, incluindo
a função das células imunes circulantes e a liberação de citocinas. Esses
processos são conduzidos pela regulação de várias vias intermediárias,
incluindo síntese de NO mediada por NOS induzível e atividade de COX2. Na
vasculatura, os receptores de esteróides estão presentes tanto no endotélio
quanto no músculo liso vascular e, sob condições fisiológicas, potencializam a
resposta às catecolaminas circulantes e à angiotensina II. Além disso, as
rápidas ações celulares dos esteroides podem promover o aumento das
concentrações de segundos mensageiros, como inositol-3-fosfato e AMPc.
Evidências limitadas sugerem que a insuficiência de corticosteroides
relacionada à doença crítica pode se desenvolver em estados de choque. As
causas desta insuficiência incluem insuficiência relativa do eixo hipotálamo-hipofiso-adrenal
(HPA), insuficiência adrenal ou necrose e em alguns casos resistência
periférica aos corticosteroides. Esses fatores podem se combinar para exacerbar
a disfunção vascular no choque e fornecer um mecanismo para o benefício
proposto da administração de esteroides exógenos visando reduzir a gravidade ou
duração da dependência de vasopressores no choque séptico. A insuficiência
vasopressínica (deficiência de vasopressina endógena), é tido como outro
mecanismo associado. A vasopressina atua por meio de receptores V1 específicos
na superfície das CMLV para promover o aumento do cálcio intracelular via
receptores acoplados à proteína G e fosfolipase C, que por sua vez impulsiona a
contração. No choque séptico, as concentrações plasmáticas de vasopressina
aumentam nos estágios iniciais do choque; no entanto, após 24 h, os níveis caem
para níveis abaixo do normal, o que pode ser um mecanismo para a perda do tônus
vascular. Isso pode estar associado a uma redução no número de receptores
periféricos, um fenômeno observado em modelos animais. Além disso, os receptores
V2 nas células endoteliais podem provocar vasodilatação através do aumento da
síntese de NO.
O
choque refratário, clinicamente é caracterizado
pela falta de PAM adequada e sustentada, apesar do aumento das doses de um ou mais
vasopressores[44]. Este
estado é uma combinação de um conjunto complexo de alterações fisiológicas que
se juntam, incluindo, mas não se limitando a, fluxo microcirculatório alterado,
hiperpolarização da membrana, relaxamento celular e reatividade vascular (Fig.
11). Ainda, pode evoluir para um estado de choque irreversível e
morte. A sepse é uma das principais etiologias de choque refratário (81%) na
UTI[45].

Fig
11. Mecanismos patogênicos que levam ao choque vasodilatador
refratário. ATP adenosina trifosfato, cGMP monofosfato
cíclico de
guanosina, COX-2 ciclooxigenase-2, PGI2 prostaglandina
I2, espécies reativas de oxigênio ROS[46].Uma
explanação sobre choque refratário e sua progressão para choque irreversível
pode ser encontrada neste Blog (http://blogdeterapiaintensiva.blogspot.com/2019/02/uma-visao-diferente-do-choque_2.html)
[1]
Wieruszewski, P.M., Khanna, A.K. Vasopressor Choice and Timing in Vasodilatory
Shock. Crit Care 26, 76 (2022). https://doi.org/10.1186/s13054-022-03911-7
[2] https://www.uptodate.com/contents/definition-classification-etiology-and-pathophysiology-of-shock-in adults?search=vasoplegic%20shock&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1
[3]
Vincent JL, De Backer D. Circulatory shock. N Engl J Med. 2013;369:1726–34.
[4]
Walsh M, Devereaux PJ, Garg AX et al (2013) Relationship between intraoperative
mean arterial pressure and clinical outcomes after noncardiac surgery: toward
an empirical definition of hypotension. Anesthesiology 119:507–515
[5]Khanna
AK, Maheshwari K, Mao G, et al. Association between mean arterial pressure and
acute kidney injury and a composite of myocardial injury and mortality in
postoperative critically ill patients. Crit Care Med. 2019;47:910–7.
[6] Maheshwari K,
Nathanson BH, Munson SH, et al. The relationship between ICU hypotension and
in-hospital mortality and morbidity in septic patients. Intensive Care Med.
2018;44:857–67.
[7] TAYLOR, J.G.; BISOGNANO, J. D.;
Baroreflex stimulation in antihypertensive treatment. Curr. Hypertens. Rep., v.
12(3), p.176-81, 2010.
[8] THRASHER, T. N.; Arterial baroreceptor
input contributes to long-term control of blood pressure. Curr. Hypertens.
Rep., v. 8(3), p. 249-54, 2006.
[9] Chapter 7. Vascular Control. In: Mohrman
DE, Heller L. eds. Cardiovascular Physiology, 8e. McGraw Hill; 2014. Accessed
May 07, 2022. https://accessmedicine.mhmedical.com/content.aspx?bookid=843§ionid=48779655
[10]
Porto Ribeiro Thais, Mecanismos de sinalização endotelial envolvidos na
atividade cardiovascular do α-terpineol.
Tese apresentada ao Programa de Pós-Graduação em Produtos Naturais e Sintéticos
Bioativos/UFPB como requisito parcial para obtenção do título de Doutor em
Produtos Naturais e Sintéticos Bioativos - Farmacologia. João Pessoa. 2012.
[11]
Thomas D. Giles, MD. Intrinsic Regulation of Vascular Tone: Perspectives on the
Pharmacologic Modulation of the System. https://www.medscape.org/viewarticle/574355
[12]
RIBEIRO, Thaís Porto. Mecanismos de sinalização endotelial envolvidos na
atividade cardiovascular do α-terpineol.
2012. 211 f. Tese (Doutorado em Produtos Naturais e Sintéticos Bioativos) -
Universidade Federal da Paraíba, João Pessoa, 2012.
[13]
Regulação cardíaca e da Pressão arterial. Universidade Federal Fluminense. http://webquest.sites.uff.br/regulacao-cardiaca-e-da-pressao-arterial/
[14] Guyton & Hall Physiology Review
(Guyton Physiology) 14th Edition 2021.
[15]
https://pt.wikipedia.org/wiki/Receptores_adren%C3%A9rgicos
[16] Christopher B. Overgaard and
Vladimír Džavík. Inotropes and Vasopressors. Review of Physiology and Clinical
Use in Cardiovascular Disease. Circulation. 2008;118:1047–1056
[17] https://teachmephysiology.com/urinary-system/regulation/the-renin-angiotensin-aldosterone-system/
[18] https://en.wikipedia.org/wiki/Vasopressin_receptor
[19]
Dünser MW, Takala J, Brunauer A, Bakker J. Re-thinking resuscitation: leaving
blood pressure cosmetics behind and moving forward to permissive hypotension
and a tissue perfusion-based approach. Crit Care. 2013;17:326.
[20]
Garrard CS, Kontoyannis DA, Piepoli M. Spectral analysis of heart rate
variability in the sepsis syndrome. Clin Auton Res. 1993;3:5–13.
[21] Landry DW, Levin HR, Gallant EM, et al. Vasopressin
deficiency contributes to the vasodilation of septic shock. Circulation.
1997;95:1122–5.
[22] Holmes CL, Patel BM, Russell JA et al
(2001) Physiology of vasopressina relevant to management of septic shock. Chest
120(3):989–1002
[23] Landry DW, Levin HR, Gallant EM et al
(1997) Vasopressin deficiency contributes to the vasodilation of septic shock.
Circulation 95(5):1122–1125
[24] Mederle K, Schweda F, Kattler V, et al.
The angiotensin II AT1 receptor-associated protein Arap1 is involved in
sepsis-induced hypotension. Crit Care. 2013;17:R130.
[25] Bucher M, Hobbhahn J, Kurtz A. Nitric
oxide-dependent down-regulation of angiotensin II type 2 receptors during
experimental sepsis. Crit Care Med. 2001;29:1750–5.
[26] Richard E. Gilbert e Andrew Advani.
Vasoactive Molecules and the Kidney. Capítulo do livro Brenner and Rector's The
Kidney. Eleventh Edition
[27]
https://en.wikipedia.org/wiki/Bradykinin_receptor
[28]
Kim Powell, in xPharm: The Comprehensive Pharmacology Reference, 2007
[29]
http://www.turkupetcentre.net/petanalysis/target_histamine.html
[30] Ebeigbe Anthony B. and Talabi Olufunke O.
Vascular Effects of Histamine. Niger. J. Physiol. Sci. 29(June 2014) 007
–010
[31]
Seminário apresentado pelo aluno FRANCISCO DE OLIVEIRA CONRADO na disciplina BIOQUIMICA
DO TECIDO ANIMAL, no Programa de Pós-Graduação em Ciências Veterinárias da Universidade
Federal do Rio Grande do Sul, no primeiro semestre de 2010. Professor
responsável pela disciplina: Félix H. D. González.
[32]
Oliveira MAB, et al. - Modes of induced cardiac arrest: hyperkalemia and
hypocalcemia - Literature review Rev Bras Cir Cardiovasc 2014;29(3):432-6
[33]https://www.cvphysiology.com/Blood%20Flow/BF008#:~:text=Hypoxia%3A,the%20production%20of%20vasodilator%20metabolites.
[34] Lambden, S., Creagh-Brown, BC, Hunt, J.
et ai. Definições e fisiopatologia do choque vasoplégico. Crit Care 22,
174 (2018). https://doi.org/10.1186/s13054-018-2102-1
[35]
Sepse. Caso clínico e revisão. MedicinaNet. https://www.medicinanet.com.br/conteudos/revisoes/5755/sepse.htm
[36]
Sepse. Revisão. MedicinaNet.
https://www.medicinanet.com.br/conteudos/acp-medicine/5797/sepse.htm
[37]
https://pt.wikipedia.org/wiki/CD14
[38]
Aguiar, Bibiana Butkus de. Estudo do polimorfismo -260C>T no promotor do
gene CD14 e a expressão de mCD14 e sCD14 em pacientes sépticos e em voluntários
saudáveis. Tese de Mestrado. 2005. https://repositorio.pucrs.br/dspace/handle/10923/1390
[39]
Assreuy, Jamil. Translocação nuclear de NF-KB e de receptores de
glicocorticoides em células musculares lisas: envolvimento do óxido nítrico e
do peroxinitrito. Dissertação (mestrado) - Universidade Federal de Santa
Catarina, Centro de Ciências Biológicas, Programa de Pós-Graduação em
Farmacologia, Florianópolis, 2011. https://repositorio.ufsc.br/handle/123456789/95760?show=full
[40]
Burgdorff AM, Bucher M, Schumann J. Vasoplegia in patients with sepsis and
septic shock: pathways and mechanisms. J Int Med Res. 2018 Apr;46(4):1303-1310.
doi: 10.1177/0300060517743836. Epub 2018 Jan 14. PMID: 29332515; PMCID:
PMC6091823.
[41]
Levy B, Fritz C, Tahon E, Jacquot A, Auchet T, Kimmoun A. Vasoplegia
treatments: the past, the present, and the future. Crit Care. 2018 Feb
27;22(1):52.
[42]
Omar S, Zedan A, Nugent K. Cardiac vasoplegia syndrome: pathophysiology, risk
factors and treatment. Am J Med Sci. 2015;349(1):80-8.
[43]
Shaefi S, Mittel A, Klick J, Evans A, Ivascu NS, Gutsche J, et al. Vasoplegia
after cardiovascular procedures — pathophysiology and targeted therapy. J
Cardiothorac Vasc Anesth. 2018 Apr;32(2):1013-22.
[44]
Jentzer JC, Vallabhajosyula S, Khanna AK, Chawla LS, Busse LW, Kashani KB. Management
of refractory vasodilatory shock. Chest. 2018;154:416–26.
[45]
De Backer D, Biston P, Devriendt J et al (2010) Comparison of dopamine and
norepinephrine in the treatment of shock. N Engl J Med 362:779–789
[46]
Wieruszewski, P.M., Khanna, A.K. Vasopressor Choice and Timing in Vasodilatory
Shock. Crit Care 26, 76 (2022). https://doi.org/10.1186/s13054-022-03911-7
Nenhum comentário:
Postar um comentário